


Related Flashcards
Related Topics





Cards In This Set
| Front | Back |
|
ENDOCRINE PANCREAS
|
ISLETS OF LANGERHANS--SECRETE HORMONES GLUCAGON AND INSULIN--4 CELLS THAT SECRETE HORMONES INTO BLOOD:1. ALPHA CELLS: GLUCAGON2. BETA CELLS: INSULIN3. DELTA CELLS: SOMATOSTATIN (GHIH): WHICH DECREASES GASTRIC SECRETIONS AND EMPTYING, INHIBITS RELEASE OF GASTRIN, INSULIN AND GLUCAGON4. F CELLS: PACREATIC POLYPEPTIDE (REGULAR PANCREATIC SECREATION INDUCED BY HYPOGLYCEMIA)
|
|
INSULIN
|
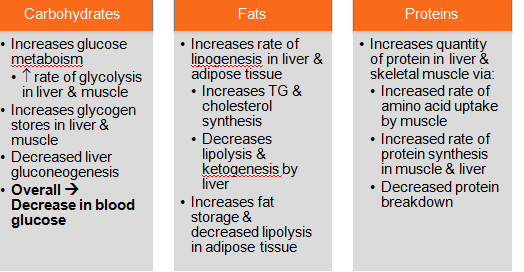 -SECRETION IS PROMOTED BY INCREASED BLOOD GLUCOSE LEVELS: HYPERGLYCEMIA -FACILITATES RATE OF GLUCOSE UPTAKE INTO CELLS OF BODY -ANABOLIC HOMRONE THAT INDUCES SYNTHESIS OF PROTEINS, LIPIDS, AND NUCLEIC ACIDS -MITOGENIC, MEANING IT PROMOTES DNA SYNTHESIS SUCH AS GROWTH AND DIFFERENTIATION -TRIGGERS THAT RELEASE INSULIN ARE: 1. INCREASED LEVELS OF GLUCAGON2. INCREASED CORTISOL LEVELS3. INCREASED LEVELS OF CERTAIN AMINO ACIDS4. VAGAL STIMULATION 5. CONDITIONS ASSOCIATED WITH INCREASED BLOOD INSULIN LEVELS AND DECREASED PERIPHERAL TISSUE INSULIN RECEPTOR BINDING (OBESITY, HIGH DIETARY CARB INTAKE, CORTISOL)6. CONDITIONS ASSOCIATED WITH DECREASED BLOOD INSULIN LEVELS AND INCREASED PERIPHERAL TISSUE INSULIN RECEPTOR BINDING (EXERCISE AND FASTING) 1. IN RESPONSE TO HIGH GLUCOSE LEVELS, GLUCOSE ENTERS BETA CELLS BY GLUCOSE TRANSPORTER2. GLUSOE IS PHOS AND METABOLIZED TO ATP3. ATP CLOSES K+ CHANNELS DEPOLARIZING CELL4. INDUCES CA2+ CHANNELS TO OPEN TRIGERRING INSULIN RELEASE5. BIPHASIC RELEASE: 1) STORED INSULIN 2) NEW SYNTHESIZED INSULIN FUNCTION:1. PROMOTES STORAGE OF INGESTED NUTRIENTS2. DECREASES BLOOD GLUCOSE LEVELS BY INCREASING GLUCOSE TRANSPORTERS ON SKELETAL MUSCLE AND ADIPOSE TISSUE--GLUT-4: INSULIN-DEPENDENT GLUCOSE TRANSPORTER--INSULIN-GLUT 4 COMPLEX-->UP TO 20 TIMES INCREASE IN GLUCOSE TRANSPORT RATE--GLUT 1-3 AND 5 GLUCOSE TRANSPORTERS ARE USED BY OTHER TISSUES THAT DO NOT USE GLUT-4: -GLUT-2 MAIN TRANSPORTER INTO BETA CELLS AND LIVER CELLS -RELATIVE LOW AFFIRMITY FOR GLUCOSE--ONLY TRANSPORTS WHEN PLASMA LEVELS ARE HIGH AFTER MEALS -GLUT-1 PRESENT IN ALL TISSUES SUCH AS NERVOUS SYSTEM GLUT-4 INSULIN DEPENDENT GLUCOSE TRANSPORTER:--GLUT 4-SKELETAL MUSCLE AND ADIPOSE TISSUE CELLS--SEQUESTERED INSIDE MEMBRANE (NON FUNCTIONAL)--INSULIN BINDING: SIGNALS MOVMENT OF GLUT 4 INTO CELL MEMBRANE WHERE IT FACILITATES GLUCOSE ENTRY INSULIN RECEPTOR BINDING:INSULIN RECEPTOR=2 SUBUNITS (ALPHA-->INSULIN BINDING, BETA-->KINASE DOMAIN) INSULIN BINDING CAUSES AUTOPHOSPHORYLATION OF BETA SUBUNIT-->TYROSINE KINASE ACTIVITY-->SIGNALING CASCADE |
|
GLUCAGON
|
LIVER IS TARGET ORGAN OF GLUCAGON (ALPHA CELLS)--TRAVELS THROUGH PORTAL CIRCULATION (LIKE INSULIN)--PROMOTES GLUCOGENESIS AND GLYCOGENOLYSIS -INCREASES BLOOD GLUCOSE
IT CAUSES DECREASED LIPOGENESIS, INCRESAED LIPOLYSIS, KETOGENESIS, PROTEOLYSIS SOMATOSTATIN (GHIH) (DELTA CELLS)--POLYPEPTIDE HORMONE, 14 AA, SHORT HALF LIFE--INVOLVEMENT IN REGULATING ALPHA AND BETA CELL SECRETIONS--DECREASES GI ACTIVITY, INCREASES USE OF ABSORBED NUTRIENTS TRIGGERS THAT RELEASE GLUCAGON:PROMOTED BY DECREASED BLOOD GLUCOSE LEVELS: HYPOGLYCEMIA SUCH AS BETWEEN MEALS, EXERCISE, STARVATION, DECREAED INSULIN, ETC |
|
HORMONES THAT AFFECT BLOOD GLUCOSE LEVELS
|
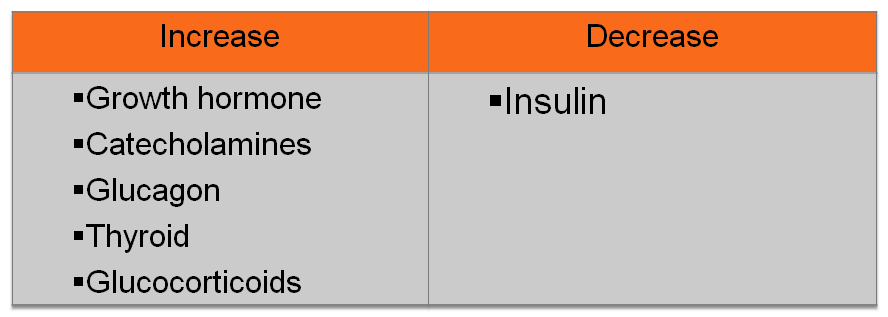 |
|
DIABETES MELLITUS
|
METABOLIC DISEASE CHARACTERIZED BY HYPERGLYCEMIA DUE TO LACK OF INSULIN PRODUCTION OR SENSITIVITY
DISORDER OF CARBOHYDRATE, LIPID, AND PROTEIN METABOLISM TYPE 1: INSULIN DEPENDENTTYPE 2: NON INSULIN DEPENDENTSECONDARY DMGESTATIONAL DM DIAGNOSTICS-POLYDIPSIA AND POLYURIA-WEIGHT LOSS (IDDM ONLY)-OBESITY (NIDDM ONLY)-FASTING VENOUS BLOOD GLUCOSE >140MG/DL=7.8MM -ORAL GLUCOSE TOLERANCE TEST (OGTT)*PERFORMED IN BORDERLINE CASES---IMPAIRED GLUCOSE TOLERANCE -GIVEN 75G ORAL GLUCOSE LOAD, AFTER 2 HOUR, BLOOD GLUCOSE >140MGDL OR ANYTIME >200MG/DL --GLYCOHEMOGLOBIN (hbA1c test) -MEASURES NON ENZYMATIC GLYCOSYLATION OF HEMOGLOBIN -INDICATES SUCCESS OF LONG TERM BLOOD GLUCOSE CONTROL |
|
TYPE 1 DIABETES MELLITUS
|
-INSULIN DPENDENT DIABETES MELLITUS-JUVENILE ONSET DM, KETOSIS PRONE DM
-PRIMARY DIABETES-->PATHOLOGY RELATED TO PRODUCTION OR PERIPHERAL INSULIN ACTION -ABRUPT ONSET OF SIGNS AND SYMPTOMS UNDER AGE 20 POSSIBLE ETIOLOGY:GENETIC SUSCEPTIBILITYINFECTIONS THAT TRIGGER AUTOIMMUNITYOTHER AUTOIMMUNE DISEASES THREE SUBTYPESTYPE 1=AUTOIMMUNE DISEASETYPE 1A-reflects environmental factors+genetics (HLA-DR4)-triggered by environmental agents, such as pancreotropic viral infection -neoantigen or molecular mimicracy-autoimmune attack on beta cells (eg: lymphocyte infiltration, islet cell destruction, autoantibodies in blood) TYPE 1B-DIFFERENT GENETIC FACTORS THAT MAKE PERSON SUSCEPTIBLE TO AUTOIMMUNE ATTACK (HLA-DR3) -CHEMICALLY INDUCED SIGNS AND SYMPTOMSSIGNS AND SYMPTOMS ARISE FROM SEVERE DEFICIENCY OF BETA CELL INSULIN PRODUCTION DUE TO ISLET CELL DESTRUCTION BY AUTOANTIBODIES-LOW BODY MASS-LOW BLOOD INSULIN-KETOACIDOSIS-AUTOANTIBODIES PATHOGENESIS-INDUCE NON ENZYMATIC GLYCOSYLATION OF EXTRACELLULAR AND INTRACELLULAR PROTEINS (ADVANCED GLYCOSYLATION END PRODUGES) -HIGH INTRACEULLAR LEVELS OF SORBITAL IN NON GLUT4 TISSUES TREATMENT:-STABLIZE BLOOD GLUCOSE THROUGH INSULIN INJECTIONS, INSULIN INFUSION PUMPS OR EXPERIMENTAL ISLET CELL TRANSPLANTATION -DIETARY REGULATION---REGULAR MEALS AND SNACKS---RESTRICTION OF REFINED CARBOHYDRATES---LIMITED CALORIC INTAKE TO MAINTAIN IDEAL BODY WEIGHT EXERCISE (RISK OF HYPOGLYCEMIA) |
|
KETOACIDOSIS
|
INSULIN DEFICIENCY-->EPINEPHRINE-->RELEASE OF GLUCAGON
GLUCAGON:-STIMULATES GLUCOGENESIS-->HYPERGLYCEMIA-->OSMOTIC DIURESIS-->DEHYDRATION -BREAKDOWN OF ADIPOSE TISSUE-->FATTY ACIDS CONVERTED TO FATTY ACETYL Co A in liver-->OXIDIZED BY LIVER MITOCHONDRIA TO KETONE BODIES-->KETONEMIA AND KETONEURIA-->METABOLIC KETOACIDOSIS KETOACIDOSIS TRIGGERED BY:INSULIN DEFIENCIECY (eg: IDDM)LOW CARB DIET DUE TO HYPOGLYCEMIA TRIGGERING GLUCAGON RELEASESTRESS OR ILLNESS: ANTI-STRESS HORMONES (CORTICOL, EPINEPHRINE, GH) OPPOSE INSULIN AND TRIGGER GLUCAGON RELEASE MECHANISM OF KETOACIDOSIS-LACK OF INSULIN INDUCES RELEASE OF GLUCAGON RELEASE BY ALPHA CELLS-->HEPATIC EFFECTS --increased glucogenesis --increased glycogenolysis --impaired glucose utilization by liver --increased amino acid uptake by liver --increased ketone body formation (ketogenesis):incomplete oxidation of free fatty acids -ACETONE -ACETONACETIC ACID -BETA-HYDROXYBUTRYIC ACID -->KETOACIDOSIS=METABOLIC ACIDOSIS SIGNS AND SYMPTOMS:VOLUME DEPLETION (INCREASED OSMOTIC PRESSURE IN TUBULE-->OSMOTIC DIURESIS)-HYPOTENSION--------DRY SKINAND PULSE-WEAK RAPID PULSE-ODOR OF ACETONE ON BREATH-HYPERGLYCEMIA-MAY PROGRESS TO COMA-INSULIN SHOCK (HYPOGLYCEMIA DUE TO INSULIN OVERDOSE) CAN LEAD TO COMA-DISTINGUISH BETWEEN DIABETIC (KETOTIC) COMA VS. INSULIN SHOCK COMA----NO VOLUME DEPLETION, NORMAL BLOOD PRESSURE, MOIST SKIN, HYPGLYCEMIA |
|
TYPE 2 DIABETES MELLITUS
|
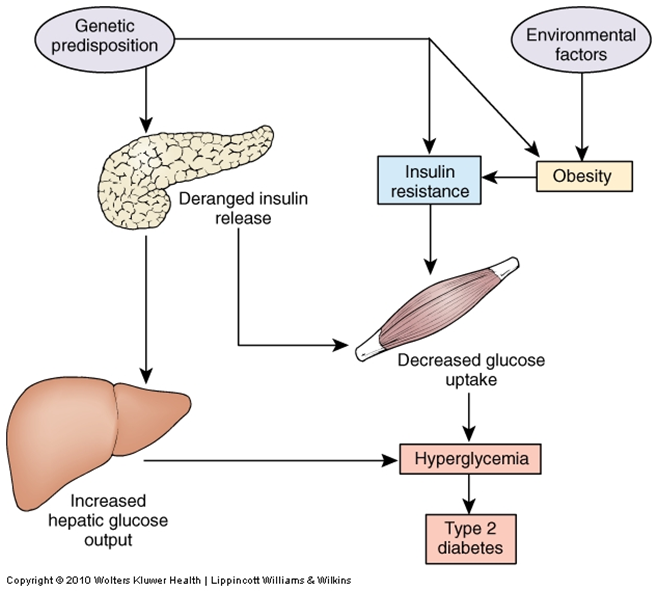 -NON INSULIN DPENDENT DIABETES, ADULTS ONSET DM (>30 YEARS) -PRIMARY DIABETES-->PATHOLOGY RELATED TO INABILITY OF PERIPHERAL TISSUES TO RESPOND TO INSULIN---LEADS TO DECREASED GLUCOSE UPTAKE AND INABILITY TO SUPPRESS GLUGOGENESIS-OBESITY AND GENETIC FACTOR-NORMAL OR INCREASED BLOOD INSULIN ETIOLOGY-GENETICALLY DEFECTIVE INSULIN RECEPTORS OR SIGNALLING-OBESITY: TRIGLYCCERIDES AND HORMONES INTERRUPT INSULIN SIGNALLING-B CELL DYSFUNCTION PATHOGENESISPERIPHERAL INSULIN RESISTANCE MECHANISM:-ABNORMAL BETA CELL INSLUIN RELEASE UPON PANCREATIC STIMULATION-DECREASED NUMBER OF CELL SURFACE INSULIN RECEPTORS-POST RECEPTOR DEFECT (INSULIN BINDING FAILS TO INDUCE INSERTION OF GLUT4 INTO MEMBRANE)-DECREASED NUMBER OF GLUT4 TRANSPORTERS SIGNS AND SYMPTOMS:-PERIPHERAL INSULIN RESISTANCE-NORMAL OR INCREASED BLOOD INSULIN-NO ISLET CELL AUTO-ANTIBODIES-NO KETOACIDSOS-CENTRAL OBESITY-GENETICS TREATMENT -EXERCISE-WEIGHT REDUCTION-DIETARY RESTRICTION (OF REFINED CARBS AND CALORIES)-ORAL HYPOGLYCEMIC OR EUGLYCEMIC AGENTS-INSULIN INJECTIONS |
|
COMPAIRE TYPE 1 AND TYPE 2 DM
|
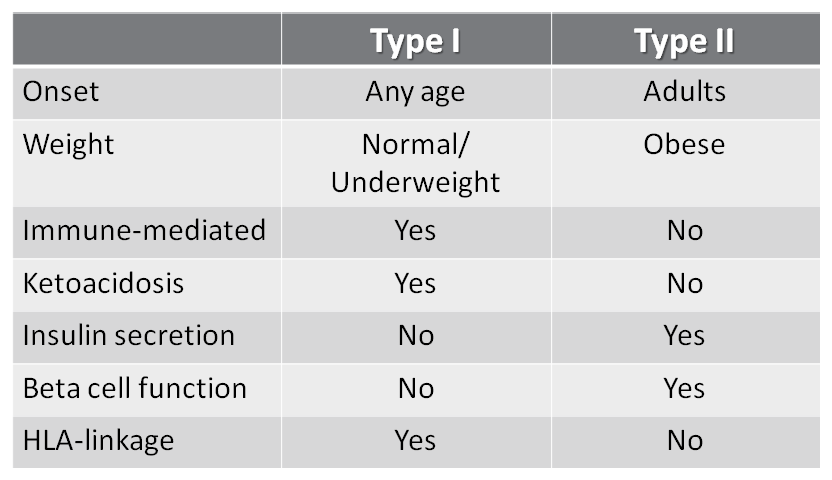 |
|
SECONDARY DIABETES MELLITUS
|
HYPERGLYCEMIA ASSOCIATED WITH IDENTIFIABLE CAUSES OF ISLET DESTRUCTION SUCH AS PANCREATITIS, SURGERY, TUMOURS, HEMOCHROMATOSIS VIA PANCREATIC DAMAGE
OR HYPERGLYCEMIA ASSOCIATED WITH DRUGS OR HYPERSECRETED HORMONES WITH ANTAGONIZE INSULIN ACTION SUCH AS GLUCOCORTICOSTERIODS, hGH, ADRENERGIC AGONISTS OR CORTISON IN CUSHING'S, GH IN ACROMEGALY, ADRENALIN |
|
GESTATIONAL DIABETES MELLITUS (GDM)
|
-DIABETES IN PREVIOUSLY NON-DIABETIC PREGNANT FEMALES
ETIOPATHOGENESIS-MAY BE DUE TO EXCESS OF A GROWTH HORMONE-LIKE HORMONE DURING PREGNANCY TO METABOLIC-STRESS OF PREGNANCY IN WOMEN WITH SUCEPTIBILITY TO NIDDM -MOST RETURN TO NORMAL POST-PARTUM BUT HALF CASES OF OBESE WOMEN WITH GDM DELVELOP NIDDM WITHIN 5-20YEARS -CANDIDATE HORMONE: HUMAN CHORIONIC SOMATO-MAMMOTROPIN (hCs)--INCREASES BREAST TISSUE TO PREPARE FOR LACTATION--INCREASES PROTEIN SYSNTHESIS-INCREASES BETA-OXIDATION AND DECREASES GLUCOSE USE -POORLY CONTROLLED DIABETES IN PREGNANT WOMEN INCREASES EMBRYO-FETUS MORBIDITY AND MORTALITY |
|
LONG-TERM COMPLICATIONS OF POORLY CONTROLLED DIABETES MELLITUS
|
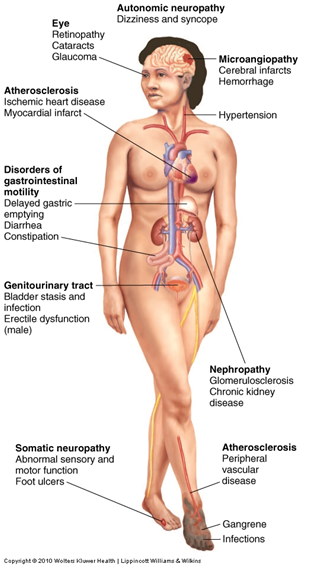 -NEPHROPHATHY-NEUROPATHY-RETINOPATHY-VASCULAR DISEASE-IMMUNODEFICIENCY-ATHEROSCLEROSIS |
|
HYPERGLYCEMIA CAUSES BLOOD VESSEL LESIONS BY 3 PATHWAYS
|
1. PROTEIN GLYCOSYLATION-GLUCOSE IN CELL FORMS BYPRODUCT THAT COMBINES AMINO GROUPS OF PROTEINS PRODUCING "ADVANCED GLYCATION END PRODUCTS" (AGE)
-AGE DESTROYS ECM, MODIFIES CIRCULATING PLASMA PROTEINS WHICH THEN DESTROY ENDOTHELIAL, MESANGIAL AND MACROPHAGE CELLS -LEADS TO ATHEROGENESIS AND MICROVASCULAR INJUY 2. INTRACELLULAR HYPERGLYCEMIA IN NON-GLUT4 CELLS -DISTURBS ANTIOXIDANT PATHWAYS AND CAUSES TISSUE DAMAGE:---DEPLETION OF NADPH-->DECREASED GLUTHIONINE (DECREASED ANTI-OXIDANT CAPACITY)---HIGH GLUCOSE-->GLUCOSE IS CONVERTED TO SORBITOL AND FRUCTOSE -SORBITOL AND FRUCTOSE INCREASE INTRACELLULAR OSMOLARITY -INFLUX OF WATER -CELL INJURY AND POSSIBLE CELL DEATH 3. ACTIVATION OF PKC -PKC IS ACTIVATED BY EXCESS CA2+ (WHICH IS ACTIVATED BY GLUCOSE): WHICH CAUSES -PROANGIOGENESIS-->RETINOPATHY-INACTIVATION OF eNOS(VASODILATOR) AND INCREASES E1 (VASOCONSTRICTOR)-INCREASES PROFIBROGENIC MOLECULES-->ECM DEPOSITION-INCREASES PROCOAGULANT MOLECULES-->VASCULAR OCCLUSIONS-INCRESAES PROINFLAMMATORY CYTOKINES |
|
CHRONIC COMPLICATIONS OF IDDM AND NIDDM
|
DIABETIC NEPHROPATHY--GLOMERULAR LESIONS -ISCHEMIC GLOMERULAR DAMAGE -GLOMERULOSCLEROSIS -THICKENING OF GBM-->LEAKINESS--PYELONEPHRITIS--END-STAGE CHRONIC RENAL FAILURE (UREMIA)
DIABETIC NEUROPATHY--CHARCTERIZED BY SCHWANN CELL INJURY, MYELIN DEGENERATION AND AXONAL DAMAGE--PRODUCING: -SYMMETRIC PERIPHERAL NEUROPATHY (DECREASED SENSATION IN LEGS, FEET, HANDS, ARMS) -AUTONOMIC NEUROPATHY EG: HYPOTENSION, BOWEL AND BLADDER DYSFUNCTION, SEXUAL IMPOTENCE, DECREASED SWEATING DIABETIC RETINOPATHY--MICROANEURYSMS-BLOOD VESSEL DAMAGE--RETINAL NEOVASCULARIZATION -PKC AND VEGF--RETINAL DETACHMENT -BLINDNESS -TREAT WITH LASER PHOTOCOAGULATION (CAUTERIZATION OF LEAKY BV'S)--GLAUCOMA -INCREASED INTRA-OCULAR PRESSURECATARACT FORMATION -OPACITIES OF CRYSTALLINE LENS--GLYCOSYLATION AND SORBITOL DIABETIC VASCULAR DISEASE:--MICROANGIOPATHY: DAMAGE TO WALLS OF SMALLEST BLOOD VESSELS 1.PURPURA 2. HYALINE ARTERIOSCLEROSIS ---CORONARY ARTERY DISEASE, CEREBRAL VASCULAR DISEASE AND PERIPHERAL VASCULAR DISEASE: -CEREBRAL ISCHEMIA, STROKES, ANGINA PECTORIS, AMI--INTERMITTENT CLAUDICATION -INADEQUATE BLOOD SUPPLY TO LOWER LIMBS--GANGRENE OF LOWER EXTREMITIES --ISCHEMIC GASTROENTEROPATHY (NECROTIC BOWEL) IMMUNODEFICIENCY--DECREASED SPECIFIC AND NON-SPECIFIC IMMUNE DEFENSES LEAD TO SUSCEPTIBILTY TO INFECTIONS--UTI-SKIN LESIONS, CELLULITIS-ORAL OR GENITAL YEAST INFECTIONS (CANDIDIASIS) --ACCELERATED ATHEROSCLEROSIS-INCREASED BLOOD TRIGLYCERIDE AND LDL CHOLESTEROL AND DECREASED HDL CHOLESTORAL (DYSLIPIDEMIAS)-INCREAED TENDENDCY OF PLATELETS TO AGGREGATE-->OCCLUSIVE EVENTS AND ATHEROMAS (ACCUMULATION/SWELLING OF ARTERIAL WALLS) |
|
DIFFERENCE BETWEEN HIGH BLOOD SUGAR AND LOW BLOOD SUGAR
|
|
