Mucopolysaccharidoses-what causes this?
-name the 6 lysosomal storage diseases |
|
-accumulation
of sulfated polysaccharides / GAGs1•Hunter syndrome2•Hurler syndrome3•Sanfilippo syndrome4•Morquio syndrome5•Maroteaux-Lamy syndrome6•Sly syndrome |
| |
Sphingolipidoses-what causes this?
-name the 3 lysosomal storage diseases |
|
-accumulation
of sphingolipid•Gaucher's disease: (accumulation
of glucocerebroside)•Niemann-Pick disease: (accumulation of sphingomyelin & cholesterol)•Gangliosidoses; Tay Sachs disease: (GM2 gangliosidosis) |
| |
|
•Mucolipidoses -what causes this?-name the 3 lysosomal storage diseases |
|
-accumulation glycoprotein & glycolipid•I : sialidosis•II: I-cell disease•III: pseudo-Hurler polydystrophy |
| |

I-cell disease (Mucolipidosis II)
|
|

•Deficiency of N-acetylglucosamine phosphotransferase
→ Absense of M6P tag
•Acid
hydrolases lacking M6P are secreted extracellularly
→
Undigested substrates accumulate as inclusion bodies, progressively damages
cells
•Skeletal
abnormalities (lack of growth)
•Coarse features
•Restricted joint
movement
•Psychomotor
retardation
•Enlarged liver,
spleen, heart valves
•Death CHF / RTI
•Life expectancy
<10yrs |
| |

Pseudo-Hurler Polydystrophy
(Mucolipidosis III)- why pseudo? |
|

-
Milder
form of I-cell
-
Site
on enzyme that is recognized by N-acetylglucosamine phosphotransferase to put
M6P tag on it is mutated
-
Still
get some tags à
some activity (lysosomal activity is totally fine otherwise)
Later onset, survival into adulthood |
| |
Sphingolipidoses: Gaucher disease - what is deficient?-Gaucher cells?-What is a key effect?
|
|
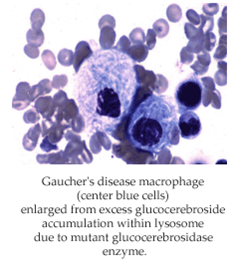
-
**most
common lysosomal storage disease**
-
Deficiency
of glucocerebrosidase à
accumulation of glucocerebroside (a glycosphingolipid)
in macrophages = Gaucher’s cells
-
progressive organomegaly with
marrow and CNS infiltration
-
splenomegaly
à inc destruction of blood
components à
anemia, neutropenia, thrombocytopenia
-
neurological
symptoms: type I- convulsions, hypertonia, MR, apnea; type II- myoclonus,
convulsions, dementia, ocular m. apraxia
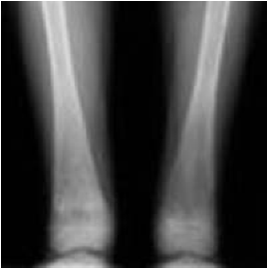
75% develop visible bony abnormalities |
| |
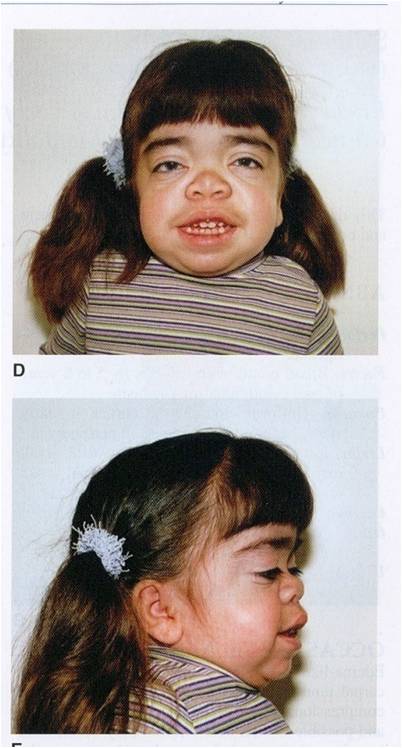
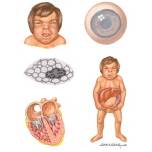
Hurler’s syndrome (MucoPS IH) -Mode of inheritance? -What is deficient? -What accumulates? -Name 5 most important symptoms
|
|
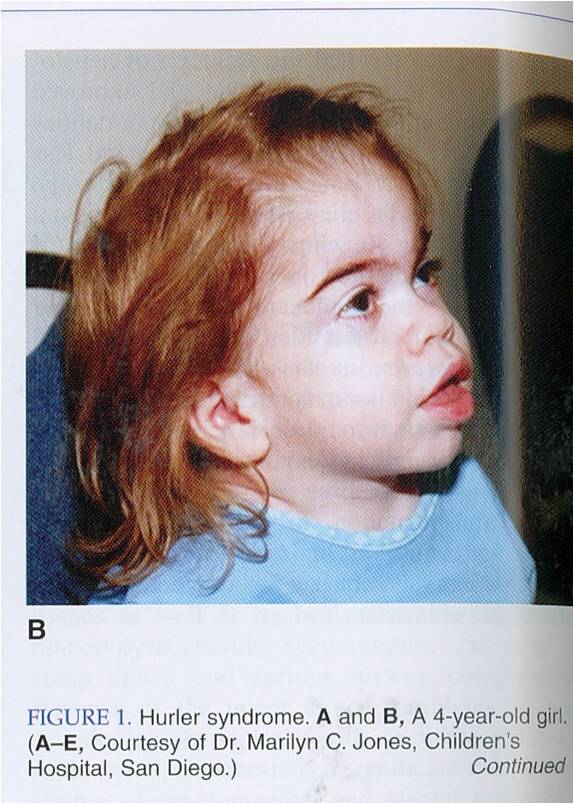
-
Most
severe MPS
-
AR
-
Deficiency
of α-L-iduronidase à
accumulation of dermatan sulfate and heparin sulfate
-
Found
in urine, cartilage, periosteum, tendons, valves, meninges, cornea
-
Initial
normal growth, but at a few months old:
-
Physical and mental deterioration
-
Organomegaly, hydrocephalus
-
Deafness
-
Hirsutism
-
à
corneal clouding!
-
Death
Scheie & Hurler-Scheie Syndrome à
milder disease
|
| |
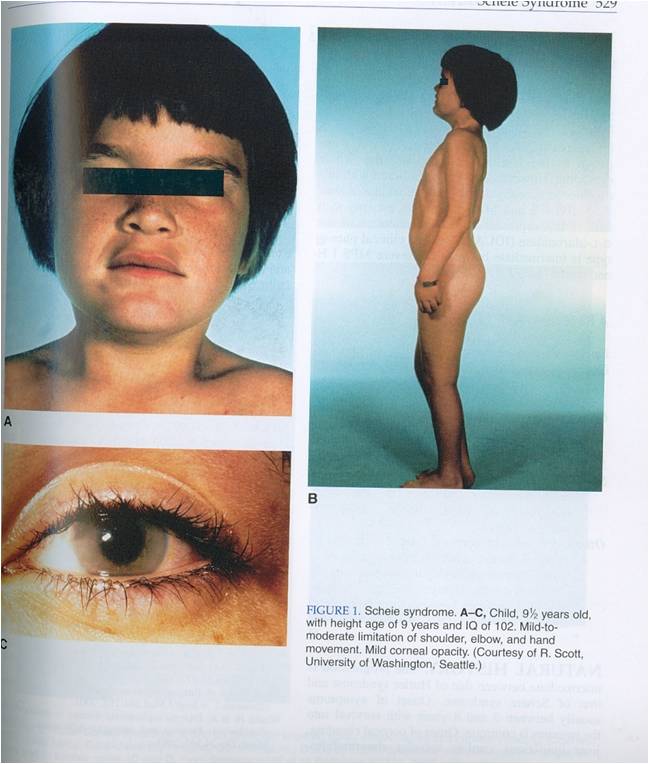
Scheie & Hurler-Scheie
syndrome
(MPS IS & MPS IHS)
|
|
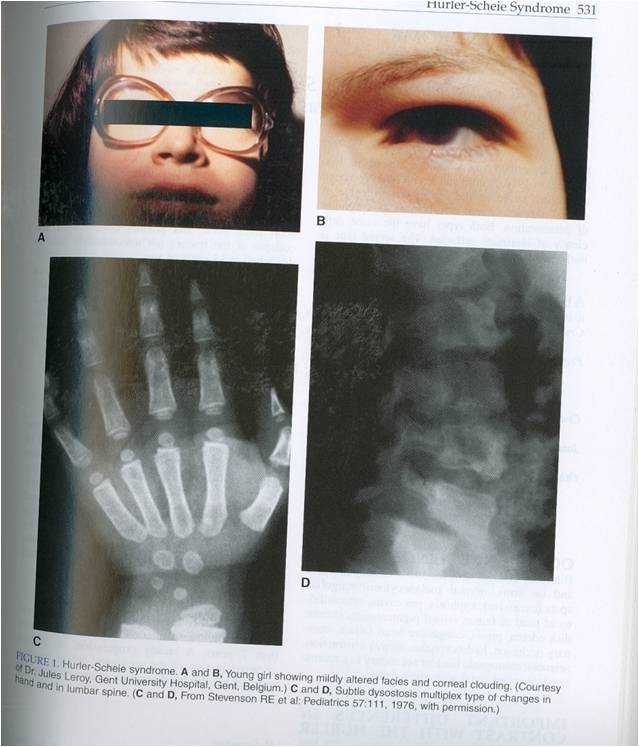
•Residual
α-L-iduronidase activity
•Milder disease (Scheie = mildest MPS I) |
| |
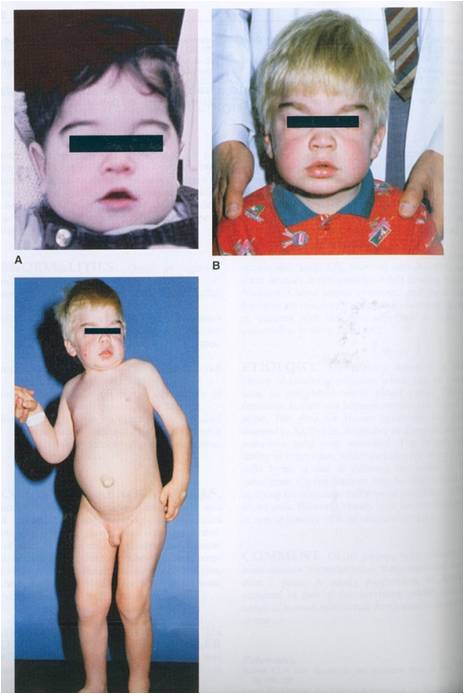
Hunter’s syndrome (MPS II)
|
|
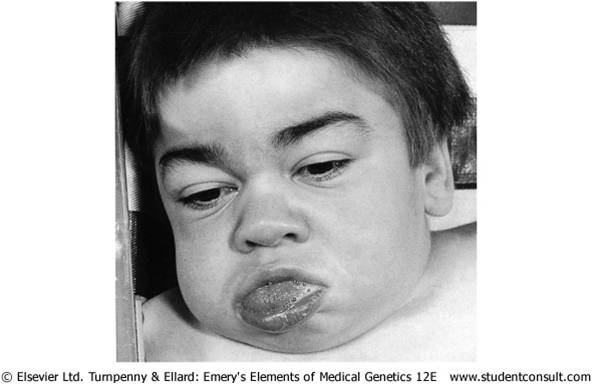
-
Deficiency
of iduronodate sulphatase
-
X-linked
-
Similar
to Hurler Syndrome, but later presentation and milder course and:
à NO
corneal clouding! |
| |
|
Because MPC Hurlers and Hunters syndromes are so similar, what can tell them apart? |
|
Hurlers: -Symptoms begin at a few months-corneal clouding-Autosomal Recessive
Hunters: -Symptoms begin at 2 yrs-no corneal clouding -x-linked
How to never forget?To be a Hunter, you need two eyes to aim for the X! |
| |

Sanfilippo
syndrome (MPS III) -What is the defect? - The effects?
|
|

-
Defect
in heparan sulphate degradation (types A-D)
-
Normal
development for first 1-2 yrs, followed by
-
Progressive
mental retardation & increasing behavioural disturbance
-
Aggressive
behaviour & destructiveness
-
Hyperactivity
-
Sleep
disturbance
(Hurler
= most severe MPS...
but children with Sanfilippo live longer with more severe behavioural problems)
|
| |
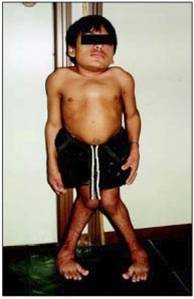
Morquio
syndrome (MPS IV) -What is defective?
|
|
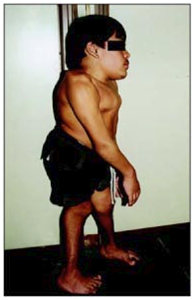
-
Defective
degradation of keratan sulphate à deficiency of galactosamine-6-sulfatase or
β-galactosidase (milder)
-
Short
stature
-
Pectus
carinatum (pigeon chest…Marfans)
Normal IQcan be confused but it is still a lysosomal storage disease |
| |
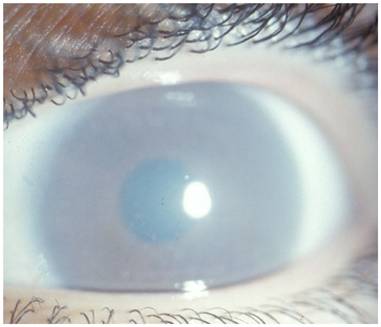
Maroteaux-Lamy syndrome (MPS VI)
|
|
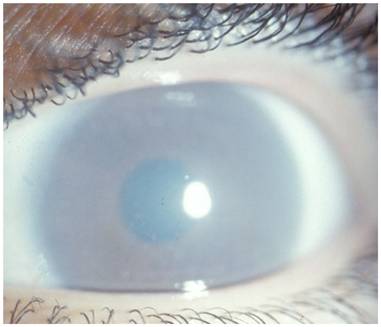
-
Deficiency
of arylsulphatase B
Similar to Hurler but normal IQ |
| |
|
Sly syndrome (MPS VII) -deficiency? |
|
-
Deficiency
of β -glucoronidase
Wide variability in severity; different mutations |
| |
Specialised lysosomes
Chédiak-Higashi syndrome
-Mode of inheritance? -Phenotype? According to prof., What is the mutation? For USMLE purposes, what is the defect?
|
|
-
Rare,
AR
-
Mutation
in CHS1/LYST, a lysosomal trafficking regulatory protein that is
normally involved in vesicle fusion (but USMLE: defect in microtubule
polymerization that causes defects in cytoplasmic granules)
-
Delayed fusion of phagosome w/
lysosome in leukocytes
-
Autophagocytosis of mealnosomes
in melanocytes à
albinism
-
Granular defects in Natural
Killer cells and platelets
-
Recurrent
infections (life threatening)
-
Hypopigmentatio -
Mild
coagulation defect -
Varying
neurological problems
Treatment: bone marrow transplant |
| |
